

I saw this paper by Chris Nosko and Steve Tadelis presented two years ago. I have wanted to blog about it in all that time but because it came out of ebay Research Labs, it was not available. Happily, that constraint has now been lifted. It is a wonderful piece of research and the application of economics.
The starting point is to think about ebay as a platform and how seller reputation impacts on it. When a seller does a poor job, this impacts both on their reputation but also on buyer’s perceptions of the platform as a whole. Thus, there is a positive externality to any individual seller maintaining a good reputation as it enhances the platform as a whole. In addition, if buyers have a bad experience and leave the platform without leaving feedback, bias occurs. Some years ago, I wrote about this issue in relation to hotel reviews but, actually, for ebay, the effects are more straightforward.
What the paper demonstrates is that these reputational externalities are not just theory but are real for ebay. Put simply, feedback is disproportionality absent for buyers that end up not returning to ebay. Just look at this graph:
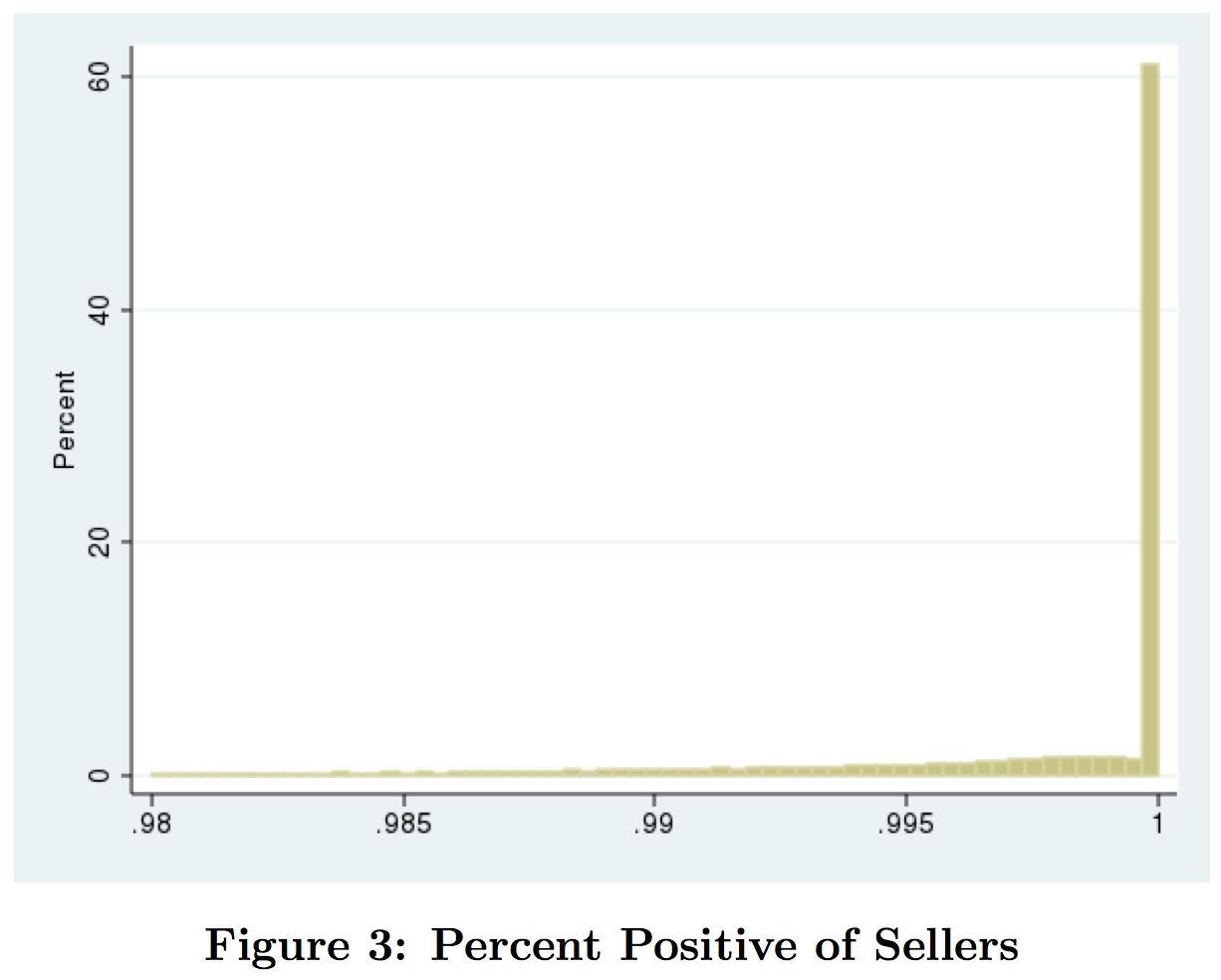
To be sure, this graph already starts at the 98th percentile! Suffice it to say, if all sellers are positive on ebay, showing us the percent positive for each seller is not going to do much in the way of sorting. Of course, the problem is that, if the reputation mechanism worked perfectly, we would see the same thing. No one would buy from anyone who didn’t happen to have a perfect rating and so no other sellers could be in business.
Of course, the paper shows that this rosy outcome is not really the case. Indeed, in just one month in October 2011 in the US there were 44 million transactions with 0.39% with negative feedback and 1% had an actual dispute ticket opened on the ebay system. Suffice it to say, that alone suggests the system isn’t working.
So what’s going on here? Basically, the authors postulate (with some anecdotal support) that it is more costly (perhaps much more costly as you can get sued or harassed) to leave negative feedback than to leave positive feedback. In other words, silence is the cheaper option for consumers. In Hirschman’s famous parlance, the system wants people to exercise voice but it pushes them towards exit. Moreover, it does so in an asymmetric fashion.
Given this hypothesis, what would be the sensible thing to do if it were true? What you would do is adjust the feedback score to take into account the share of transactions for a seller where no feedback was given. Instead of percent positive you would adjust by a seller’s propensity to receive feedback. Here is the authors’ example:
Walking through a specific example, consider two sellers, A who had 120 transactions and and B who had 150. Assume that both received one negative feedback, and 99 positive feedbacks. Using ebay’s PP measure, both have a PP of 99% ( 99 ). However, seller A had 99+1 only 20 silent transactions with no feedback while seller B had 50 silent transactions. We define “effective” PP (EPP) as the number of positive feedback divided by total transactions, in which case seller A has an EPP of 82.5% while seller B has an EPP of only 66% and is a worse seller on average. Importantly, ebay does not display the total number of transactions a seller has completed and buyers cannot therefore back-out a seller’s EPP score.
It turns out that this yields a measure where there are lots of differences amongst sellers.
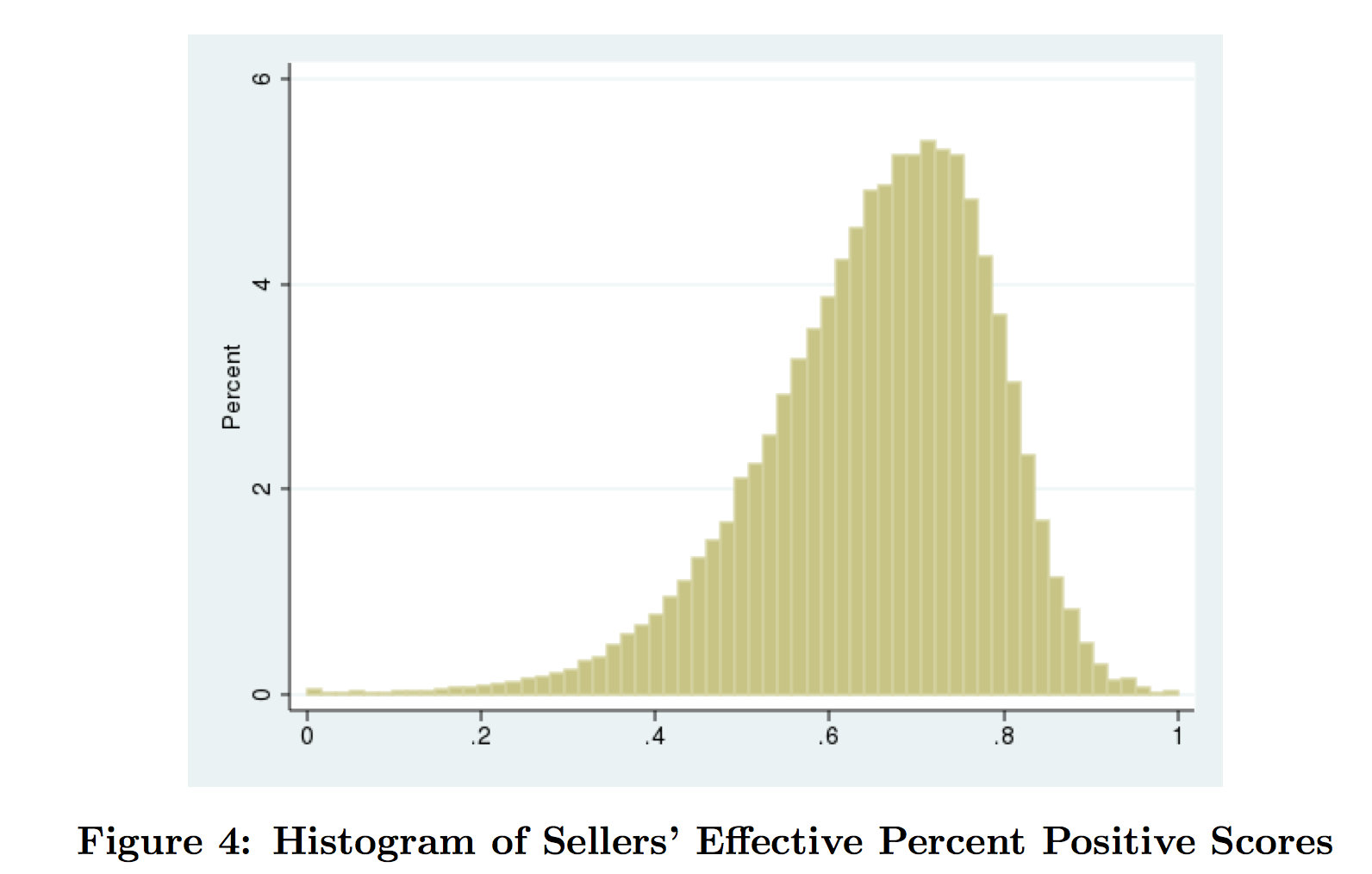
But how do you know if this new measure is any good? After all, it was based on a particular hypothesis about buyer behaviour — a hypothesis that could not be directly confirmed. The answer, of course, is to look at situations where the buyer did give negative feedback or did open a dispute. This was actually 3.39% of the transactions in the authors’ dataset. They show that when buyers had an observably bad experience, the seller was more likely to have a lower adjusted reputational score and vice versa. So this seemed to be the right stuff.
They took about a million buyers in 2011 and looked at the effect of the seller’s adjust score (which the buyers never got to see) in a current transaction with the buyer’s propensity to continue to be on ebay in the future. They show that their adjusted score predicts whether buyers come back better than other indicators of reputation.
Here is the where the final piece in the puzzle came together. Armed with this, because they not only had access to ebay data but access to ebay, the researchers ran an experiment. They changed ebays search ranking outcome for some buyers to reflect the adjusted score. That is, searchers were more likely to see results from higher scoring sellers. Treated buyers turned out to be more likely to return to ebay than the control group.
Now this doesn’t mean that platforms such as ebay should switch to the new rank. After all, if sellers knew this they might pressure buyers even more just to manipulate them. Moreover, the score may matter differently for different categories of transactions. Nonetheless, the paper itself is a beautiful analysis of economics done right and what you should do with big data rather than just hunting for correlations.

Thanks for posting this. It does look like a promising way to tackle a long-standing problem.
My main doubt (I confess I haven’t read the paper yet) is whether adopting the new system would change behaviour. The absence of negative reviews indicates that a social norm (courtesy) has evolved where giving positive feedback is, like signing a guest book at a B&B with a nice comment, a courtesy: a way to politely conclude a transaction.
Until now, if you have a bad experience, then giving a non-review has been a way out of the awkwardness of giving a bad review. I wonder if, once it was known that not giving a review is basically the same as giving a negative review, the number of non-reviews might fall off.
..which I guess is what you said in your final paragraph. Maybe I was wasting space.